Glossary of Bioinformatic Terms
By: Heather Blankenship and Kelsey Florek
December 18, 2019
General Bioinformatic Terms
Bioinformatics
This discipline is involved in the collection, analysis, and storage of complex biological data, including, but not limited to genetic data. The field combines computer science, statistics, and several aspects of biology to be able to understand highly complex data.
AMD (Advanced Molecular Detection)
This commonly may mean any form of sequencing or high-level molecular technology. There is an Office of Advanced Molecular Detection (OAMD) at CDC that provides support and input on many cross-cutting areas using advanced technologies
Genomics
-
Gene – sequence of DNA that encodes a region for a molecule (ie protein or enzymes) with function
-
Genotype vs Phenotype – Genotypes are the genetic make-up of an individual. Phenotypes are the physical traits and characteristics of an individual and are influenced by their genotype and the environment.
- Gene Regulation
- Cis regulation – Any molecular interaction that regulates the transcription of nearby genes on the same DNA molecule, such as the role of a gene promote
- Trans regulation – Any molecular interaction that regulates the transcription of genes on a different DNA molecule, such as a transcription factor regulating both alleles of a target gene or genes.
-
Indels – A type of DNA sequence variation marked by the insertion or deletion of nucleotides, Defined as insertion or deletion of bases in the genome of an organism. An INDEL mutation named with the blend of insertion and deletion. It refers to a length difference between two allele caused by a SEQUENCE INSERTION or by a SEQUENCE DELETION.
-
Mobile elements – sequences of DNA that have the ability to move from one organism to another, move into different locations in a genome or change the copy number in the bacteria. These element include plasmids, bacteriophages and transposable elements
-
Mutation Rates – rate in which a single nucleotide is replaced by a different nucleotide Recombination – rearrangement of genetic material. Both prokaryotes and eukaryotes can perform recombination.
-
Operon – It is a genetic regulatory system found in bacteria and their viruses in which genes coding for functionally related proteins are clustered along the DNA. This feature allows protein synthesis to be controlled coordinately in response to the needs of the cell. A typical operon consists of a group of structural genes that code for enzymes involved in a metabolic pathway, such as the biosynthesis of an amino acid. These genes are located contiguously on a stretch of DNA and are under the control of one promoter (a short segment of DNA to which the RNA polymerase binds to initiate transcription). A single unit of messenger RNA (mRNA) is transcribed from the operon and is subsequently translated into separate proteins.
- Structural variants (SVs) – DNA sequence variants that are 50bp or larger, including insertions, deletions, inversions, duplications and translocations
Programming Languages
-
BASH / Shell Script – A programming language that is directly used by the Linux operating system. It allows for direct control of the operating system functions.
-
Command line / Command based – The command line allows the user to interact with their computer without graphical user interface (GUI) or mouse. The command line is essential to perform bioinformatics analysis. It allows the user to manipulate files and programs in ways that are not possible using a GUI.
-
Linux – An operating system much like Windows or OSX. The field of bioinformatics relies heavily on Linux-based computers and software. Although some bioinformatics programs can be compiled to run on Mac OS X and Windows systems, many require the Linux environment to run properly.
-
PERL – Another programming language similar to Python.
-
Python – Python is a popular programming language object often used for scientific computing. The Biopython utilize python-based software for bioinformatics research, thereby creating high quality reusable modules and scripts.
Sequencing Instruments and Chemistry
-
PacBio – It is a single molecule real time (SMRT) sequencing developed by Pacific Biosciences, also referred as PacBio sequencing. Unlike second generation sequencing, PacBio sequences each base-pair individually. Currently, this method is widely used to study larger genomes, transcriptome metagenomics and epigenetics.
-
Tape station – The Agilent 2100 Bioanalyzer (Tape Station) is a microfluidics-based platform for size determination, quantification and quality control of DNA and RNA.
-
Pyrosequencing – In pyrosequencing, the sequencing reaction is monitored through the release of the pyrophosphate during nucleotide incorporation. A single nucleotide is added to the sequencing chip which will lead to its incorporation in a template dependent manner. This incorporation will result in the release of pyrophosphate which is used in a series of chemical reactions resulting in the generation of light. Light emission is detected by a camera which records the appropriate sequence of the cluster. Any unincorporated bases are degraded by apyrase before the addition of the next nucleotide. This cycle continues until the sequencing reaction is complete.
-
Paired end sequencing –
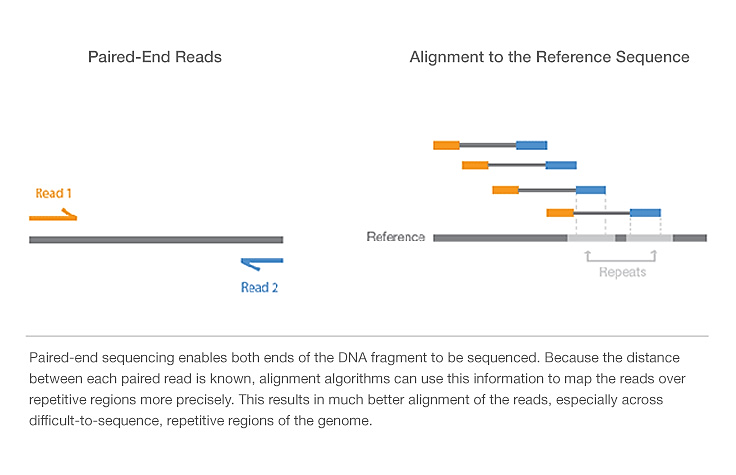
It involves sequencing both ends of the DNA fragment and generate high quality sequence data. It’s a simple work flow allows generation of unique ranges of insert sizes.
-
Long read sequencing – Third generation sequencing also known as long read sequencing (LRS). LRS allows for the retrieval of much longer (>10,000bp) sequencing reads than widely-used short read sequencing systems (75-300bp). Long-read sequencing (LRS) has become increasingly important due to its strengths in resolving complex DNA regions as well as in determining full-length RNA molecules. Two important LRS technologies have been developed during the past few years, including single-molecule, real-time sequencing by Pacific Biosciences, and nanopore sequencing by Oxford Nanopore Technologies.
-
Short read sequencing – Next generation sequencing also known as short read sequencing. The technology utilizes highest output platform continue to have relatively short read lengths on the order of 35-300 bases per read. The amazing increase in data output and decrease in cost per base sequenced has been driven primarily short-read sequencing technologies such as the Illumina and Ion Torrent platforms.
-
Massively parallel sequencing – Massively parallel sequencing (MPS) technology, also referred to as next-generation sequencing, is a high-throughput approach to DNA sequencing in which miniaturized, parallelized platforms are used for sequencing 1 million to 43 billion short reads per instrument run. Next-generation sequencing (NGS) is transforming the landscape of clinical microbiology and public health laboratories. The applications of NGS are wide-ranging and include whole-genome sequencing, microbiome analysis/metagenomics, transcriptome profiling, infectious disease diagnosis, pathogen discovery, and public health surveillance.
-
Base Calling – Base calling is the process of assigning nucleotides or bases (ATGC) with chromatogram peaks. Next generation sequencing platforms, which use fluorescently labeled reversible terminators have a unique color for each base. These are incorporated in to the complementary strand of the DNA template and captured with a sensitive CCD camera. These images are processed into signals which are used to infer the order of nucleotides also referred as base calling. The accuracy is, measured by a Q score (Phred quality score), a common metric to assess the accuracy of a sequencing run.
-
Adapter – They are oligonucleotides bound to 5’ and 3’ end of each fragment in a sequencing library. Adapters include platform-specific sequences for fragment recognition by the sequencer. For example, the P5 and P7 sequences enable library fragments to bind to the flow cells of Illumina platforms.
-
Barcode sequences – Multiplex sequencing allows large numbers of libraries to be pooled and sequenced simultaneously during a single run on a high-throughput Illumina NextGen sequencer. Individual “barcode” sequences are added to each DNA fragment during next-generation sequencing (NGS) library preparation so that each read can be identified and sorted out based on the fingerprinting of the genome prior to final data analysis.
-
Sanger Sequencing – DNA sequencing method that relies on a primer to bind to the DNA sequence of interest to amplify the region of interest, sequences a single DNA fragment
Bioinformatic Analysis
-
ANI (average nucleotide identity) – provides a measure of how similar two genomes are at the nucleotide level
-
Coverage – number of reads that are aligned at a specific nucleotide
-
Contig (contiguous DNA) – sequences of DNA that have been assembled together as the result of overlapping sequences into a consensus sequence and resulting in a contiguous sequence
- De novo assembly – assembly of sequencing reads without using a reference sequence as a guide
- DNA Sequence Analysis
-
CgMLST (Core genome MLST) – examines only the genes that are shared among all of the strains to assign alleles, these genes are shared by the majority of the strains within a genus
-
MLST (Multi Locus Sequence Typing) – traditionally used 6-12 housekeeping genes and Sanger sequencing to identify gene variants and alleles, which can be used to generate sequence types (ST), standardized allele schemes
-
WgMLST (whole genome MLST) – examines all genes within the genome to assign allele codes to an organism, examined on a gene by gene basis and can be either the whole gene or part of the gene to form the locus, variants within the locus are given new allele codes to generate wgMLST types.
-
Consensus – sequence of nucleotides that represents the most commonly found base pair at a specific position, identified by aligning the raw reads Reference assembly: mapping reads to a reference genome in order to assembly the reads into contigs or closed genomes
-
Reference genome – a consensus sequence of DNA for an organism
-
Variants – sequences of DNA that differ at specific positions when the sequences are aligned
- Alleles – variant of a sequence, variant does not have to affect the phenotype
-
SNP (single nucleotide polymorphism) – a nucleotide position that can be used to discriminate strains in a population
-
HqSNP (High quality SNP) – utilizes a program such a lyve-SET to identify SNPs that have at least 20X coverage and uses a high quality reference genome
- Query sequence – BLAST (Basic Local Alignment Search Tool) uses statistical methods to compare a DNA or protein input sequence (“query sequence”) to a database of sequences (“subject sequences”) and return those sequences that have a significant level of similarity to the query sequence.
-
- File Types
-
FASTQ file – contains the raw sequencing data, contains the NGS read data including both the DNA sequences and the quality sequencing information for each read, each read has the read ID, the base calls, additional information and sequencing quality metrics
- FASTA file – contains DNA/RNA or protein sequences, “>” denotes the header/sequence identifier
- SAM (Sequence Alignment/Map) file – contains the alignment of reads to a reference genome
- BAM file – compressed binary version of a SAM file
-
-
Filtering – Pre processing of raw reads before assembly to remove any reads that do not meet a specified quality parameters
-
Gene or sequence annotation – Sequence annotation is the process of marking specific regions or sites of interest in the protein sequence, such as post-translational modifications, binding sites, enzyme active sites, local secondary etc. Annotation may be defined as the part of genome analysis that is customarily performed before a genome sequence is deposited in GenBank and described in a published paper.
-
k-mer – k-mer are often used in computational genomics. k-mer refer to all the possible sub-sequences of a defined length obtained from a set of sequence information.
-
Metagenomics – the study of complex bacterial communities to examine diversity of organisms at various taxonomic levels. Sequencing can be performed using 16S primers (16S sequencing) or by sequencing all DNA in a sample (shotgun sequencing) to identify genes present and identification of organisms
-
Multiple sequence alignment – Multiple Sequence Alignment (MSA) is generally the alignment of three or more biological sequences (protein or nucleic acid) of similar length. In contrast, Pairwise Sequence Alignment tools are used to identify regions of similarity that may indicate functional, structural and/or evolutionary relationships between two biological sequences. ClustalW is widely used program for multiple sequence alignment.
-
NCBI (National Center for Biotechnology Information) – part of the National Laboratory of Medicine and National Institute of health. The Entrez Global Query Cross Database search system is widely adopted by NCBI for array of nucleotide and protein sequences, protein structures, PubMed, taxonomy, whole genome etc.
-
N50 – A weighted average length; specifically, the N50 length is the length such that 50% of the genome has been assembled into contig or scaffold sequences of this length or longer.
-
Pipeline – the chain of processing data and moving files through different processes or functions to produce the desired files and results, also referred to as a workflow
-
Proteomics – the study of all the proteins in a system and their interactions, structure and composition
-
Q Score – the quality score is the probability of an incorrect base call; Q30: 1 in 1000 times incorrect, thus, each nucleotide is 99.9% accurate
-
Reading frame and Open reading frame (ORF) – An ORF is a sequence of DNA that starts with start codon “ATG” and ends with any of the three termination codons (TAA, TAG, TGA). Depending on the starting point, there are six possible ways (three on forward strand and three on complementary strand) of translating any nucleotide sequence into amino acid sequence according to the genetic code. These are called reading frames.
-
Reads – fragments of sequenced DNA base pairs that are generated during sequencing, the raw data from a sequencing machine
-
Sequence Similarity – It is a method of searching sequence databases by using alignment to a query sequence. Similarity of two sequences refers to the degree of match between corresponding positions in sequence.
- Source Control / Version Control – tracking of changes made to files or authorship to be able to recall previous versions. Github is a way to keep track of changes made to files and identify who made the changes.